Sede del Ministerio de Sanidad.
El Gobierno ha aprobado el
Real Decreto que regula la evaluación de tecnologías sanitarias en España, una norma que ordena el sistema encargado de analizar
medicamentos, productos sanitarios, tecnologías digitales, procedimientos clínicos y modelos organizativos antes de su financiación, incorporación, modificación de uso o eventual desinversión en el Sistema Nacional de Salud.
El texto crea un Sistema de Evaluación de Tecnologías Sanitarias con un
Consejo de Gobernanza y dos oficinas diferenciadas, una para
medicamentos, integrada en la Aemps, y otra para
tecnologías no farmacológicas, asumida por la RedETS.
La norma
fija plazos, metodologías comunes, garantías de independencia, participación de pacientes y profesionales, uso de datos en vida real y transparencia, aunque deja claro que las evaluaciones no serán vinculantes, sino que servirán para informar las decisiones posteriores de financiación, precio y cartera de servicios.
El Real Decreto completo, a continuación:
11587 Real Decreto 415/2026, de 27 de mayo, por el que se regula la evaluación de tecnologías sanitarias.
El desarrollo y la incorporación a la práctica clínica de las tecnologías sanitarias constituye un elemento básico de la protección de la salud de las personas que, a su vez, aspiran a obtener oportunamente un beneficio de ellas cuando se enfrentan a problemas de salud que no están adecuadamente resueltos por las alternativas disponibles. Sin embargo, el desarrollo y el acceso a las tecnologías sanitarias tiene un importante impacto social que excede el ámbito exclusivo de la salud, ya que son una fuente de investigación, innovación y conocimiento, así como un motor del desarrollo industrial, de la creación de empleo, y del crecimiento económico.
El objeto de este real decreto es regular las actividades de evaluación de las tecnologías sanitarias (en adelante, ETS) en tanto estén dirigidas específicamente a informar decisiones de la administración competente relativas a la incorporación, financiación, precio, reembolso, modificación de las condiciones de uso o desinversión en tecnologías sanitarias.
Las tecnologías sanitarias incluyen medicamentos, productos sanitarios, pruebas para el diagnóstico in vitro, procedimientos clínicos, terapias y productos sanitarios digitales, modelos organizativos y medidas para la prevención, el diagnóstico o el tratamiento de enfermedades. Cada una de estas tecnologías tiene una regulación propia y unos requerimientos diferentes para ser aplicadas en la práctica clínica que aseguran la garantía de la calidad, la seguridad y la eficacia.
La ETS es un proceso científico basado en datos contrastados que permite determinar la eficacia relativa de tecnologías sanitarias existentes o nuevas. Por ello, se trata de una herramienta necesaria para informar las decisiones de las autoridades competentes estatales y autonómicas en el diseño e implementación de la cartera de prestaciones sanitarias con el objeto de promover un sistema sanitario equitativo, eficiente y de alta calidad. Es importante hacer notar que la evaluación de tecnologías sanitarias informa la toma de decisiones, pero no constituye la propia toma de decisiones.
Para cumplir con sus fines, la evaluación de cualquier tecnología sanitaria comprende tanto los aspectos clínicos como los aspectos no clínicos de la misma. Se han identificado nueve ámbitos para la ETS, de los que cuatro son clínicos y cinco son no clínicos. Los cuatro ámbitos clínicos son la identificación de un problema de salud y la tecnología sanitaria actual, el análisis de las características técnicas de la nueva tecnología sanitaria, su seguridad relativa y su eficacia clínica relativa. Los cinco ámbitos de evaluación no clínicos se refieren al coste y la evaluación económica de una tecnología, y a sus aspectos éticos, organizativos, sociales y jurídicos, tal y como se recoge en los considerandos del Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021, sobre evaluación de las tecnologías sanitarias y por el que se modifica la Directiva 2011/24/UE. Dentro de los aspectos no clínicos, se incluye adicionalmente también el ámbito ambiental y es probable que puedan crecer en el futuro conforme se evalúen más terapias y productos sanitarios digitales.
Por otra parte, el procedimiento para la financiación pública de los medicamentos y productos sanitarios para su inclusión en la prestación farmacéutica debe tener en cuenta criterios generales, objetivos y publicados, según el artículo 92 del texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios, aprobado por el Real Decreto Legislativo 1/2015, de 24 de julio. Estos criterios se refieren a dimensiones que están incluidas en la esfera de la ETS y, en concreto y entre otros, al «valor terapéutico y social del medicamento y beneficio clínico incremental del mismo teniendo en cuenta su relación coste-efectividad» o el «valor social del producto sanitario y beneficio clínico incremental del mismo teniendo en cuenta su relación coste-efectividad». Además, es necesario tener «en cuenta el componente de innovación, para avances terapéuticos indiscutibles por modificar el curso de la enfermedad o mejorar el curso de la misma, el pronóstico y el resultado terapéutico de la intervención y su contribución a la sostenibilidad del Sistema Nacional de Salud si, para un mismo resultado en salud, contribuye positivamente al Producto Interior Bruto». Por otro lado, para tecnologías sanitarias distintas de los medicamentos, la Ley 16/2003, de 28 de mayo, de cohesión y calidad del Sistema Nacional de Salud, establece en el artículo 21.2 que «las nuevas técnicas, tecnologías o procedimientos serán sometidas a evaluación, con carácter preceptivo y previo a su utilización en el Sistema Nacional de Salud, por la Red Española de Agencias de Evaluación de Tecnologías Sanitarias y Prestaciones del Sistema Nacional de Salud» (en adelante, RedETS).
En este sentido, la ETS debe proporcionar la información necesaria, de acuerdo con su carácter científico, que permita a los órganos decisores estatales y autonómicos el cumplimiento de las funciones que vienen determinadas en la legislación con respecto a la incorporación de prestaciones en el sistema público. En consecuencia, deben identificarse claramente ambos ámbitos de trabajo, de evaluación y de decisión, con sus respectivas estructuras, competencias y responsabilidades.
Cualquier desarrollo nacional relacionado debe estar alineado con el citado Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021. Este reglamento establece un modelo de evaluación conjunta de los cuatro dominios pertenecientes al ámbito clínico para medicamentos y productos sanitarios. El informe de evaluación clínica conjunta resultante deberá tenerse debidamente en cuenta por los Estados miembros.
Al mismo tiempo, este reglamento no impide que los Estados miembros realicen los análisis clínicos complementarios que sean necesarios para el conjunto de su proceso nacional de ETS ni restringe la facultad de los Estados miembros de realizar evaluaciones no clínicas.
Por tanto, la ETS debe guardar continuidad y coherencia con la evaluación conjunta europea y tiene que estar orientada a las fases posteriores de toma de decisión sobre financiación y precio, acceso y despliegue en el sistema sanitario, abarcando su ciclo de vida completo. Es muy importante garantizar que el proceso de ETS sea independiente, participativo, transparente, adaptativo, vinculado a una decisión oportuna y adaptado a las características de cada tecnología, por un lado, y de las personas destinatarias, por otro. Para ello es imprescindible que se garantice la participación de las organizaciones de pacientes y consumidores y, con ellos, la de las personas con discapacidad, enfermedades raras u otros grupos específicos cuyas necesidades deben ser atendidas con esa especificidad, incluyendo también a las personas cuidadoras. Además, ha de tenerse en cuenta que, para medicamentos, las evaluaciones clínicas conjuntas solo se realizan sobre aquellos autorizados para su comercialización por procedimiento centralizado o sobre nuevas indicaciones de medicamentos de los que ya se dispone de una evaluación clínica conjunta, y que dichos informes, de manera general, deben limitarse a la descripción de la eficacia clínica relativa sin juicio de valor ni categorización de los resultados en la salud. Sin embargo, el proceso completo de la ETS va más allá porque es necesario establecer el valor clínico añadido teniendo en cuenta el grado de innovación y el valor para pacientes, familias y personas cuidadoras, y todo ello siguiendo el principio de buena práctica administrativa, aspirando a lograr los máximos niveles de calidad, transparencia e independencia. En definitiva, la ETS, como emana del Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021, debe garantizar un denominador común de equidad en la UE, pero respetando al mismo tiempo las competencias y capacidad de gestión según las atribuciones de los Estados miembros, incorporando la valoración y posicionamiento de cada tecnología para una mejor gestión de la provisión de salud a la ciudadanía en un marco de eficiencia y sostenibilidad.
La evaluación de medicamentos, productos sanitarios u otro tipo de tecnología sanitaria ha tenido un recorrido diferente en España y otros países europeos. Si bien las industrias de ambos tipos de tecnología son esenciales para la salud, tienen características que las hacen notablemente distintas, y su desarrollo y condiciones para su puesta en mercado obedecen a regulaciones diferentes. Además, la experiencia nos ha enseñado que, aunque el marco conceptual de la ETS es muy similar, la demanda y ritmo de incorporación al Sistema Nacional de Salud de los diferentes tipos de tecnología sanitaria es distinto. Para dar un desarrollo de la ETS adaptado a todo tipo de tecnología, es importante seguir manteniendo una troncalidad común pero una configuración separada para medicamentos, por un lado, y para tecnologías sanitarias no farmacológicas, por otro.
Esta diferente configuración es la que se ha adoptado en el Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021, pero también es la que se ha venido manteniendo en España con la participación de la Agencia estatal Agencia Española de Medicamentos y Productos Sanitarios (en adelante, la AEMPS), por un lado, y la RedETS, por otro, de acuerdo con sus competencias estatutarias. Ambas instituciones han realizado sus informes siguiendo diferente metodología, aunque sirviendo al mismo objetivo de informar las decisiones relativas a la cartera de prestaciones y financiación pública.
Por consiguiente, es necesario adoptar una metodología común y coordinada que dé continuidad a lo dispuesto en el Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021, y amplíe el alcance de la evaluación al incluir aspectos de competencia exclusiva de los Estados miembros como la categorización del efecto relativo, la evaluación económica complementaria y finalmente el posicionamiento de la tecnología, para lo que se habrá de considerar adicionalmente el resto de ámbitos no clínicos incluyendo aspectos éticos, sociales, organizativos, jurídicos y ambientales. Esta metodología también podrá ser aplicada a las tecnologías fuera del alcance del reglamento si así fuera necesario.
A tal efecto, este real decreto desarrolla el denominado «Sistema para la evaluación de las tecnologías sanitarias» que se despliega a través de las «Oficinas para la evaluación de las tecnologías sanitarias», constituidas por la AEMPS para los medicamentos y por la RedETS para las tecnologías sanitarias no farmacológicas. Asimismo, es necesario asegurar la continuidad con las decisiones mediante la participación de los actores relevantes, incluyendo las comunidades autónomas, en el órgano para la gobernanza del sistema y en su continuidad mediante las estructuras para la adopción de las tecnologías, que intervienen inmediatamente antes de la toma de decisiones y en las que participan también las autoridades autonómicas.
Este real decreto se ajusta a los principios de buena regulación a los que se refiere el artículo 129 de la Ley 39/2015, de 1 de octubre, del Procedimiento Administrativo Común de las Administraciones Públicas. A estos efectos, los principios de necesidad y eficacia se justifican en las razones de interés general descritas en los párrafos precedentes, constituyendo el instrumento más adecuado para garantizar la consecución de las metas propuestas. La norma es acorde con el principio de proporcionalidad, ya que contiene la regulación imprescindible para atender las necesidades identificadas, no existiendo otras alternativas menos restrictivas o que impongan menos obligaciones para lograr los objetivos fijados. La norma no impone cargas administrativas innecesarias o accesorias y racionaliza, en su aplicación, la gestión de los recursos públicos. En relación con el principio de transparencia, los objetivos de la norma están claramente definidos y justificados en este preámbulo y se ha posibilitado la participación activa de los destinatarios en la elaboración de la norma de acuerdo con el artículo 26 de la Ley 50/1997, de 27 de noviembre, del Gobierno, y el artículo 7 de la Ley 19/2013, de 9 de diciembre, de transparencia, acceso a la información pública y buen gobierno, mediante un trámite de consulta pública previa a la elaboración del texto, en primer lugar, y el trámite de audiencia pública correspondiente, con la participación, respectivamente, de 43 y 74 asociaciones o particulares de los sectores potencialmente afectados por esta norma.
Asimismo, el desarrollo es plenamente congruente con el ordenamiento jurídico europeo y nacional y, en concreto, el Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021, la Ley 16/2003, de 28 de mayo, de cohesión y calidad del Sistema Nacional de Salud y el texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios, aprobado por el Real Decreto Legislativo 1/2015, de 24 de julio, redundando en una mayor seguridad jurídica, con una mayor precisión en la definición y alcance de los diferentes actos, lo que dota de coherencia y estabilidad al marco normativo en esta materia.
De acuerdo con lo dispuesto en la Ley 16/2003, de 28 de mayo, de cohesión y calidad del Sistema Nacional de Salud, este real decreto ha sido objeto de informe previo por parte del Comité Consultivo y del Pleno del Consejo Interterritorial del Sistema Nacional de Salud. Además, la autoridad administrativa independiente Agencia Española de Protección de Datos ha emitido su informe, en virtud de lo dispuesto en la Ley Orgánica 3/2018, de 5 de diciembre, de Protección de Datos Personales y garantía de los derechos digitales, y en el Estatuto de la citada Agencia, aprobado por el Real Decreto 389/2021, de 1 de junio. En el proceso de elaboración de esta norma se ha consultado, entre otros, a las comunidades autónomas, a las ciudades de Ceuta y Melilla y a los sectores afectados.
De conformidad con lo dispuesto en el artículo 149.1.16.ª de la Constitución, este real decreto se dicta de acuerdo con la competencia exclusiva que ostenta el Estado en materia de bases y coordinación general de la sanidad y de legislación sobre productos farmacéuticos.
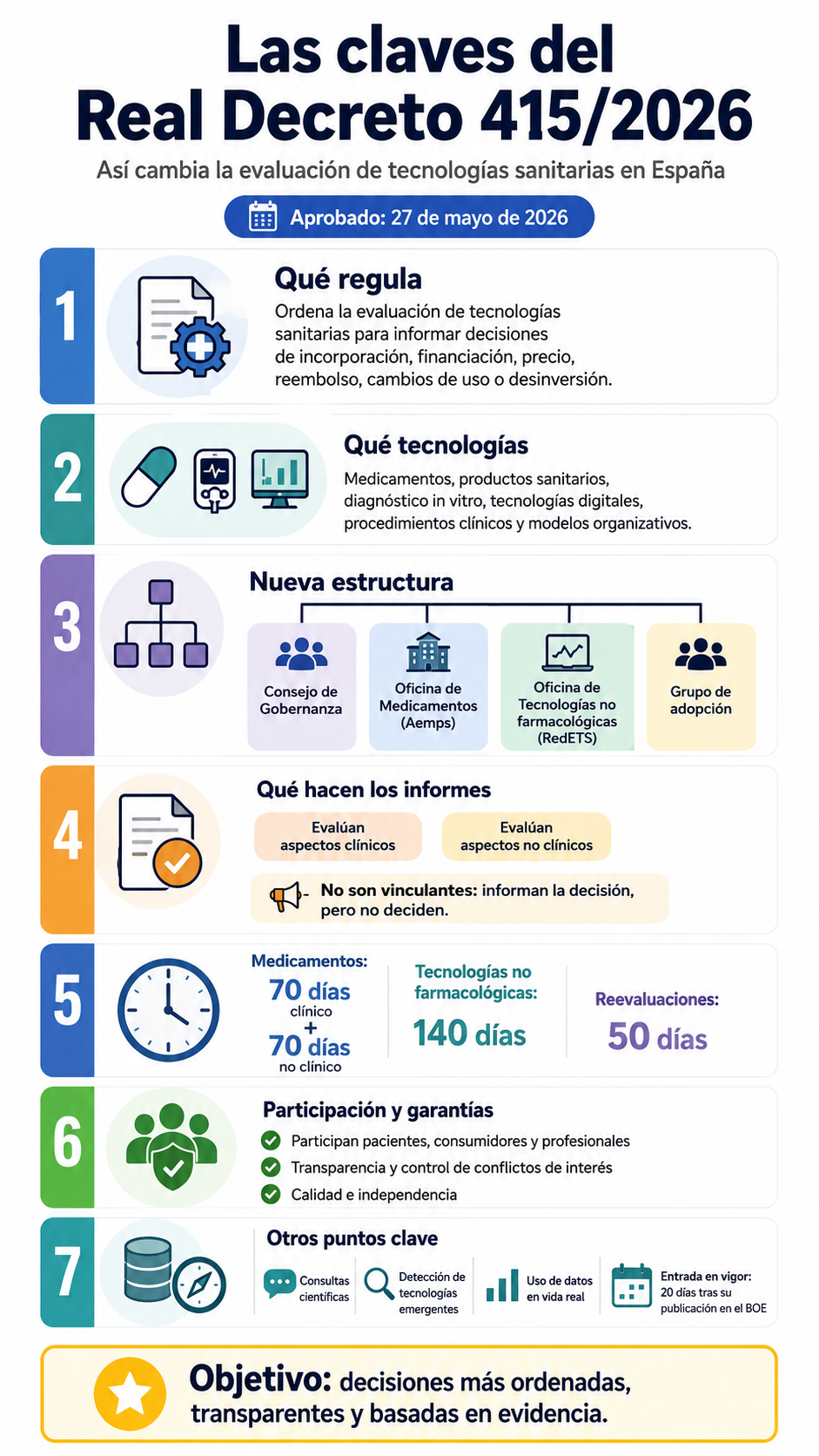
Las claves del Real Decreto de Evaluación de Tecnologías Sanitarias.
|
En su virtud, a propuesta de la Ministra de Sanidad, con la aprobación previa del Ministro para la Transformación Digital y de la Función Pública, de acuerdo con el Consejo de Estado, y previa deliberación del Consejo de Ministros en su reunión del día 26 de mayo de 2026,
DISPONGO:
CAPÍTULO I
Disposiciones generales
Artículo 1. Objeto.
1. Este real decreto regula las actividades de evaluación de tecnologías sanitarias (en adelante, ETS) en tanto estén dirigidas específicamente a informar decisiones de la administración competente relativas a la incorporación, financiación, precio, reembolso, modificación de las condiciones de uso o desinversión en tecnologías sanitarias, o formen parte del despliegue de dichas decisiones en la práctica clínica. A tal efecto, sus objetivos son:
a) Desarrollar un marco de la ETS coherente y complementario, en todo lo que le es de aplicación, con el Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021, sobre evaluación de las tecnologías sanitarias y por el que se modifica la Directiva 2011/24/UE.
b) Desarrollar un marco, normas y metodologías comunes para la ETS a nivel nacional en aquellos aspectos que son competencia exclusiva de los Estados miembros.
c) Desarrollar los mecanismos de coordinación necesarios que permitan trasladar de manera eficiente el conjunto de evaluaciones a las decisiones de las administraciones competentes.
2. Están excluidas del ámbito de aplicación de este real decreto aquellas actividades de ETS que no tengan como razón de ser la toma de decisiones informadas por parte de la administración.
Artículo 2. Definiciones.
A los efectos de lo dispuesto en este real decreto se entiende por:
a) «tecnología sanitaria»: un medicamento, producto sanitario, o procedimientos médicos o quirúrgicos, así como las medidas para la prevención, el diagnóstico o el tratamiento de enfermedades utilizados en la asistencia sanitaria;
b) «evaluación de las tecnologías sanitarias»: lo definido en el artículo 2, apartado 5, del Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021;
c) «ámbitos de evaluación clínicos»: se refiere a la identificación de un problema de salud y la tecnología sanitaria que se utiliza actualmente para su manejo, el examen de las características técnicas de la tecnología sanitaria objeto de evaluación, su seguridad relativa y su eficacia clínica relativa;
d) «ámbitos de evaluación no clínicos»: se refieren a los costes, utilización de recursos, la evaluación de la eficiencia y del impacto presupuestario de una tecnología sanitaria, así como a sus aspectos éticos, organizativos, sociales, jurídicos y ambientales;
e) «medicamento»: toda sustancia o combinación de sustancias según se define en el artículo 1, punto 2, de la Directiva 2001/83/CE del Parlamento Europeo y del Consejo, de 6 de noviembre de 2001, por la que se establece un código comunitario sobre medicamentos para uso humano;
f) «producto sanitario»: según se define en el artículo 2, punto 1, del Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, sobre los productos sanitarios, por el que se modifican la Directiva 2001/83/CE, el Reglamento (CE) n.º 178/2002 y el Reglamento (CE) n.º 1223/2009 y por el que se derogan las Directivas 90/385/CEE y 93/42/CEE del Consejo;
g) «producto sanitario para diagnóstico in vitro»: un producto sanitario para diagnóstico in vitro según se define en el artículo 2, punto 2, del Reglamento (UE) 2017/746, del Parlamento Europeo y del Consejo, de 5 de abril de 2017, sobre los productos sanitarios para diagnóstico in vitro y por el que se derogan la Directiva 98/79/CE y la Decisión 2010/227/UE de la Comisión;
h) «tecnología sanitaria no farmacológica»: incluye las tecnologías definidas en los apartados f) y g) más todas aquellas tecnologías sanitarias que no cumplan la definición de e);
i) «desarrollador de tecnologías sanitarias»: la entidad que desarrolla, produce o introduce una tecnología sanitaria y que actúa como titular o representante de dicha tecnología ante las administraciones públicas en los procedimientos de evaluación, financiación o incorporación de su tecnología en el Sistema Nacional de Salud;
j) «evaluación clínica comparada»: proceso multidisciplinar que incluye el análisis comparativo de los datos clínicos disponibles sobre una tecnología sanitaria en comparación con otra u otras tecnologías o procedimientos existentes, de conformidad con un ámbito de evaluación representativo de su posible utilización en el marco del Sistema Nacional de Salud. Se trata, por tanto, de una evaluación del valor clínico añadido o beneficio clínico adicional de la nueva tecnología;
k) «análisis económico y presupuestario»: proceso sistemático por el que se analiza la eficiencia de una tecnología para determinar los recursos agregados que exige incorporarla, en relación con los resultados en salud obtenidos con su adopción en contraste con uno o varios comparadores.
CAPÍTULO II
Organización general de la evaluación de las tecnologías sanitarias
Artículo 3. El Sistema para la evaluación de las tecnologías sanitarias.
El Sistema para la evaluación de las tecnologías sanitarias (en adelante, Sistema de ETS) está formado por el Consejo de Gobernanza (en adelante, Consejo de ETS) y dos oficinas independientes para la evaluación de las tecnologías sanitarias: la Oficina para la evaluación de los Medicamentos y la Oficina para la evaluación de las tecnologías sanitarias no farmacológicas (en adelante, las Oficinas).
Las evaluaciones generadas por el Sistema de ETS llegarán a los correspondientes órganos competentes en materia de decisiones sobre incorporación, financiación, precio, reembolso, modificación de las condiciones de uso o desinversión en tecnologías sanitarias a través del «Grupo para la adopción de las tecnologías sanitarias» (en adelante, Grupo de adopción).
Artículo 4. Consejo de Gobernanza.
1. El Consejo de ETS es el órgano de gobernanza, de los previstos en el artículo 15.2 de la Ley 40/2015, de 1 de octubre, de Régimen Jurídico del Sector Público, que supervisa y garantiza la alineación del Sistema de ETS con las políticas farmacéuticas y de prestación de servicios del Ministerio de Sanidad. El Consejo de ETS se integra en la Secretaría de Estado de Sanidad del Ministerio de Sanidad.
2. El Consejo de ETS está formado por:
a) La persona titular de la Secretaría de Estado de Sanidad, que asume la presidencia;
b) la persona titular de la Dirección General de Cartera Común de Servicios del Sistema Nacional de Salud y Farmacia, que asume la secretaría del Consejo de ETS, con voz y voto;
c) dos vocalías, con un rango mínimo de subdirector o subdirectora general, nombrados por la Dirección General de Cartera Común de Servicios del Sistema Nacional de Salud y Farmacia, uno en el ámbito de los medicamentos y otro en el ámbito de las tecnologías sanitarias no farmacológicas;
d) una vocalía, designada por la Secretaría General de Salud Digital, Información e Innovación del Sistema Nacional de Salud;
e) cuatro vocalías, designadas por las Oficinas, dos por la AEMPS y dos por la RedETS;
f) seis vocalías de comunidades autónomas designadas a propuesta del Consejo Interterritorial del Sistema Nacional de Salud, tres en el ámbito de los medicamentos y tres en el ámbito de las tecnologías sanitarias no farmacológicas;
g) dos vocalías en representación de profesionales sanitarios, nombrados por la Secretaría de Estado de Sanidad;
h) una vocalía especialista en economía de la salud, designada por la Secretaría de Estado de Sanidad;
i) una vocalía en representación de las organizaciones o asociaciones de pacientes y otra en representación de las organizaciones de consumidores, designadas por la Secretaría de Estado de Sanidad, en el segundo caso a propuesta del Consejo de Consumidores y Usuarios;
j) tres vocalías, en representación de la Administración General del Estado no pertenecientes al Ministerio de Sanidad, designadas a propuesta, respectivamente, del Ministerio de Hacienda, del Ministerio de Economía, Comercio y Empresa y del Ministerio de Industria y Turismo.
En caso de ausencia, enfermedad u otra causa legal, las vocalías del Consejo de ETS serán sustituidas temporalmente por las personas que designe la persona titular del órgano que propuso su nombramiento, hasta la reincorporación de la persona titular de la vocalía o el nombramiento de uno nueva.
Asimismo, podrán ser convocadas a las reuniones del Consejo, en función de los asuntos a tratar, personas representantes de otros departamentos ministeriales de la Administración General del Estado no pertenecientes al Ministerio de Sanidad, que participarán con voz, pero sin voto.
3. Las personas que formen parte del Consejo de ETS, a excepción de las personas nombradas por las Oficinas, no podrán participar directa ni indirectamente en las evaluaciones de las tecnologías sanitarias.
4. Las funciones del Consejo de ETS son:
a) Adoptar un reglamento interno de funcionamiento y actualizarlo cuando sea necesario; este reglamento contendrá previsiones sobre la periodicidad de sus reuniones;
b) adoptar los programas anuales de trabajo y los informes anuales de las organizaciones que forman parte del Sistema de ETS;
c) adoptar orientaciones estratégicas respecto de la ETS;
d) aprobar, a propuesta de las Oficinas o los Grupos de adopción de las tecnologías sanitarias regulados en la disposición adicional primera del presente real decreto, las directrices metodológicas de carácter no normativo para el trabajo del Sistema de ETS, garantizando su alineación con la política farmacéutica y de prestaciones del Ministerio de Sanidad, con los criterios que reglamentariamente configuren la toma de decisiones y con las recomendaciones estándar sobre la evaluación basada en la evidencia;
e) adoptar la planificación y programación de trabajo del Sistema de ETS para que sean acordes con los plazos establecidos en la normativa aplicable;
f) promover la integración y coherencia entre los diferentes actores participantes en el Sistema de ETS, así como la cooperación con los organismos pertinentes a escala nacional o de la Unión Europea para facilitar la generación de elementos de evidencia adicionales necesarios para su trabajo y la adopción de decisiones;
g) velar por que se garantice la participación apropiada de las organizaciones de partes interesadas y de personas expertas en el trabajo de los grupos de trabajo en el ámbito de sus funciones;
h) promover que exista una financiación suficiente para el programa de trabajo adoptado al que se hace referencia en el apartado b);
i) supervisar que se cumplan los criterios de calidad, participación y transparencia a los que se hace referencia en los artículos 24, 25 y 26 de este real decreto.
5. Para el cumplimiento de sus funciones, el Consejo de ETS podrá contar con el asesoramiento del Comité Asesor para la Prestación Farmacéutica del Sistema Nacional de Salud u otros comités de naturaleza asesora para las prestaciones no farmacológicas del Sistema Nacional de Salud.
Artículo 5. Oficinas para la evaluación de las tecnologías sanitarias.
1. Cada una de las Oficinas está formada por todas aquellas organizaciones de la Administración que participan en la ETS, dentro de cada ámbito de la evaluación, de medicamentos en un caso y de tecnologías sanitarias no farmacológicas en el otro, con los fines contemplados en este real decreto.
2. Cada una de las Oficinas informa las decisiones de la administración competente relativas a la incorporación, financiación, precio, reembolso, modificación de las condiciones de uso o desinversión de tecnologías sanitarias, sin formar parte de las estructuras de decisión. Cada Oficina actuará con autonomía funcional respecto de la toma de decisiones, garantizando su independencia y ausencia de conflictos de interés.
3. La «Oficina para la evaluación de los Medicamentos» se configurará como una unidad funcional de la AEMPS. Corresponde a la AEMPS el ejercicio de las funciones de dicha oficina.
La AEMPS representará a España en el Grupo de Coordinación sobre Evaluación de Tecnologías Sanitarias de los Estados miembros (Member State Coordination Group on HTA, por su denominación en inglés –en adelante, Grupo de Coordinación–), y nombrará a las personas representantes en los subgrupos de trabajo que emanan del mismo, de acuerdo con el Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021, para los temas relacionados con medicamentos.
4. La «Oficina para la evaluación de las tecnologías sanitarias no farmacológicas» asume la configuración de la RedETS. La RedETS será responsable de la evaluación de las tecnologías sanitarias no farmacológicas, que incluyen productos sanitarios, tecnologías digitales, procedimientos clínicos, así como las medidas o modelos organizativos para la prevención, el diagnóstico, el tratamiento o rehabilitación de enfermedades utilizados en la asistencia sanitaria que no incluyan específicamente medicamentos.
El Ministerio de Sanidad, como organismo coordinador de RedETS, representará a España en el Grupo de Coordinación, y RedETS nombrará, de entre sus miembros, a las personas representantes en los subgrupos que emanan del mismo, de acuerdo con el Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021, para los temas relacionados con tecnologías sanitarias no farmacológicas.
5. Las funciones generales de las Oficinas son:
a) Elaborar la evaluación comparada de los ámbitos clínicos y no clínicos, de acuerdo con lo contemplado en los artículos 7 a 12 de este real decreto, que permita una adecuada valoración de las tecnologías sanitarias para la toma de decisiones;
b) coordinar y resolver las consultas científicas contempladas en el artículo 19 de este real decreto, en colaboración, si procede, con otros integrantes del Sistema de ETS;
c) llevar a cabo las actividades de detección de tecnologías emergentes contempladas en el artículo 20 de este real decreto, que permitan la planificación del Sistema Nacional de Salud a corto y medio plazo, así como poner las tecnologías identificadas a disposición del Sistema de ETS para guiar sus actividades;
d) colaborar en el Consejo de ETS en todo aquello que favorezca el cumplimiento de sus funciones;
e) proponer y, en su caso, elaborar directrices metodológicas de carácter no normativo para el trabajo de evaluación para su aprobación en el Consejo de ETS. En todo caso, se asegurará la coherencia con la ETS a nivel europeo según el Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021, y se dará cuenta tanto de la metodología en el trabajo de evaluación como de los criterios de categorización del valor clínico añadido;
f) proponer los procedimientos internos de trabajo y establecer sus fases operativas para asegurar el cumplimiento de los plazos establecidos en el artículo 14 de este real decreto;
g) presentar cada año al Consejo de ETS el programa anual de trabajo que incluya número y el tipo previstos de evaluaciones clínicas y no clínicas, alineado con el plan anual de trabajo del Grupo de Coordinación, el número previsto de consultas científicas, las actividades para la detección de tecnologías emergentes o, en su caso, las propuestas de actualización de la cartera de servicios comunes. Estas últimas podrán ser presentadas tanto por iniciativa del Ministerio de Sanidad como por las administraciones sanitarias de las comunidades autónomas, o a petición razonada de las Mutualidades de Funcionarios o de terceros interesados;
h) presentar cada año al Consejo de ETS un informe anual, en el que se proporcione información sobre el trabajo realizado el año natural anterior a su adopción;
i) cooperar con los organismos pertinentes a escala nacional o de la Unión Europea para facilitar la generación de elementos de evidencia adicionales necesarios para su trabajo y la adopción de decisiones;
j) garantizar la participación apropiada de las organizaciones de partes interesadas y de las personas expertas en el trabajo de evaluación;
k) garantizar que, en el ejercicio de sus funciones, se cumplan los criterios de calidad, participación y transparencia a que se hace referencia en los artículos 24, 25 y 26 de este real decreto, así como el principio de accesibilidad universal;
l) gestionar y elaborar las evaluaciones clínicas y las asesorías científicas conjuntas en el marco del Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021.
CAPÍTULO III
Objeto y contenido de la evaluación de tecnologías sanitarias
Artículo 6. Tecnologías objeto de la evaluación.
1. Serán objeto de evaluación con los objetivos de este real decreto:
a) Todos los nuevos medicamentos de uso humano para los que se haya presentado una solicitud de autorización de comercialización con arreglo al procedimiento centralizado y que se pretendan comercializar en España y a los que se refiere el anexo I del Reglamento (CE) n.º 726/2004 del Parlamento Europeo y del Consejo, de 31 de marzo de 2004, por el que se establecen procedimientos comunitarios para la autorización y el control de los medicamentos de uso humano y veterinario y por el que se crea la Agencia Europea de Medicamentos;
b) los nuevos medicamentos autorizados por otros procedimientos que se pretendan comercializar en España y en los que se considere necesario realizar una evaluación comparada en atención a los criterios que serán desarrollados y publicados por el Consejo de ETS;
c) nuevas indicaciones de medicamentos ya autorizados, financiados o no, de acuerdo con los criterios que serán desarrollados y publicados por el Consejo de ETS;
d) productos sanitarios clasificados en las clases IIb o III con arreglo al artículo 51 del Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, con respecto a los cuales los paneles de personas expertas pertinentes hayan emitido un dictamen científico en el marco del procedimiento de consulta de la evaluación clínica con arreglo al artículo 54 de dicho reglamento, y que hayan sido seleccionados con arreglo al apartado 2 de este artículo;
e) productos sanitarios para diagnóstico in vitro clasificados en la clase D con arreglo al artículo 47 del Reglamento (UE) 2017/746 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, para los que los paneles de expertos pertinentes hayan emitido sus opiniones en el marco del procedimiento con arreglo al artículo 48, apartado 6, de dicho reglamento, y que hayan sido seleccionados con arreglo al apartado 2 de este artículo;
f) cualquier otro producto sanitario, procedimiento médico, quirúrgico o enfermero, tecnología digital, o medidas o modelos organizativos para la prevención, el diagnóstico o el tratamiento de enfermedades utilizados en la asistencia sanitaria que sea acordado por el Consejo de ETS;
g) aquellos medicamentos o tecnologías sanitarias no farmacológicas que hayan sido evaluadas por el Sistema y que requieran una actualización en los supuestos contemplados en el artículo 17 de este real decreto;
2. Para los objetivos previstos en este real decreto, los productos sanitarios podrán ser abordados en su evaluación como tecnología en general con todos los comparadores disponibles, o bien como productos sanitarios específicos, en los casos en los que se determine.
3. Para priorizar la evaluación de los productos sanitarios incluidos en los epígrafes
d), e) y f) de este artículo, se tendrán en cuenta las prioridades del Sistema Nacional de Salud en materia de política de productos sanitarios y los criterios contemplados en el Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021:
a) Necesidades médicas no cubiertas;
b) ser el primero de su clase;
c) las posibles repercusiones para el colectivo de pacientes, la prevención de una discapacidad, la salud pública o el Sistema Nacional de Salud, incluyendo aspectos como la sostenibilidad del sistema, el impacto de la implementación o la difusión de la tecnología;
d) la incorporación de sistemas de información que utilicen inteligencia artificial, tecnologías de aprendizaje automático o algún algoritmo equivalente;
e) una dimensión transfronteriza importante;
f) un valor añadido importante a escala de la Unión Europea que haya sido marcado como una de las prioridades a nivel sanitario.
4. Para los productos mencionados en el apartado 1.f) de este artículo podrán utilizarse los criterios de priorización contemplados en la Orden SCO/3422/2007, de 21 de noviembre, por la que se desarrolla el procedimiento de actualización de la cartera de servicios comunes del Sistema Nacional de Salud.
Artículo 7. Contenido de las evaluaciones.
1. La ETS incluirá, de manera separada, un informe sobre la evaluación clínica comparada y otro sobre la evaluación de los aspectos no clínicos que se integrarán en un mismo documento.
2. Las evaluaciones clínicas no duplicarán, en ningún caso, las evaluaciones que se hayan realizado a nivel europeo conforme al desarrollo del Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021.
3. La evaluación clínica concluirá con una categorización del valor clínico añadido según directrices metodológicas no normativas propuestas por las Oficinas y aprobadas por el Consejo de ETS. En el caso de tecnologías que dispongan de una evaluación clínica conjunta por el procedimiento europeo, las Oficinas la completarán con la categorización del valor clínico añadido.
4. La evaluación no clínica aportará, además del resto de ámbitos no clínicos, el análisis económico y presupuestario necesario, según el caso, siguiendo las instrucciones normativas y las directrices metodológicas no normativas aprobadas por el Consejo de ETS.
5. Las evaluaciones realizadas por las Oficinas, de los aspectos clínicos y no clínicos, no contendrán juicios sobre la adopción de la tecnología en el Sistema Nacional de Salud o sobre las decisiones de financiación, precio o eventual inclusión en la cartera común de servicios de la tecnología evaluada. El órgano competente para la valoración final sobre la posición relativa de la tecnología sanitaria será el Grupo de adopción regulado en la disposición adicional primera del presente real decreto.
6. El Ministerio de Sanidad publicará en su portal de internet tanto las instrucciones normativas como las directrices metodológicas no normativas, así como cualquier otra información sobre el proceso a seguir por las entidades desarrolladoras en la preparación del expediente con la información necesaria para la ETS que se presentará ante las Oficinas de Evaluación por vía electrónica. Las instrucciones normativas se ajustarán en su aprobación a lo recogido en el artículo 22.2 de este real decreto.
Cuando el expediente presentado por el desarrollador no cumpla con las instrucciones normativas o las directrices metodológicas no normativas aplicables, este contará con un plazo de diez días para la subsanación. Las instrucciones y directrices metodológicas no normativas tomarán en consideración las previsiones contenidas en el artículo 10.3 del Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021.
Artículo 8. Aspectos generales de la evaluación clínica.
1. Para determinar el valor clínico añadido se tendrán en cuenta las alternativas asistenciales u otras alternativas utilizadas dentro del Sistema Nacional de Salud.
2. Para realizar el informe de evaluación clínica se definirá el ámbito de la misma que incluirá, en particular, todos los parámetros pertinentes para la evaluación en términos de:
a) La población de pacientes;
b) la intervención o intervenciones;
c) el comparador o comparadores;
d) los resultados en salud.
3. El proceso de delimitación del alcance se hará de forma participativa y tendrá en cuenta las observaciones recibidas por las comunidades autónomas, a través del grupo de adopción, así como por pacientes y personas cuidadoras, personas expertas clínicas y otras personas expertas pertinentes, además de la información proporcionada por el desarrollador de la tecnología sanitaria.
4. Las evaluaciones se basarán en el expediente que presente el desarrollador de la tecnología sanitaria con informaciones, datos, análisis y otros elementos de evidencia completos y actualizados, además de otras fuentes de información especificadas en las directrices metodológicas no normativas de las Oficinas. En el caso de productos sanitarios evaluados como tecnología en general con todos los comparadores disponibles, las evaluaciones tendrán en cuenta los datos aportados por las entidades desarrolladoras cuando estos estén obligados a presentarlos.
5. Las evaluaciones podrán poner de manifiesto el grado de incertidumbre sobre la evidencia y se podrán proponer mecanismos o fórmulas para solventarlos que, en todo caso, deberán ser pragmáticas, proporcionadas y viables, teniendo en cuenta la naturaleza de la tecnología, la enfermedad o el área de atención de que se trate y la naturaleza de los beneficios esperados.
6. Se tendrá en cuenta la perspectiva de la persona usuaria, persona con discapacidad, paciente o sus personas cuidadoras en la fase de evaluación, para informar sobre el impacto de la enfermedad en la calidad de vida relacionada con la salud, la discapacidad y las opciones terapéuticas, incluyendo también elementos como el retraso diagnóstico, la atención de la propia patología o, cuando proceda, la usabilidad o facilidad de uso, la accesibilidad o aquellas barreras provocadas por su discapacidad.
7. Los informes de evaluación clínica contendrán conclusiones sobre el valor clínico añadido global de la tecnología sanitaria evaluada, pero no recomendaciones para la posterior toma de decisiones. Este valor clínico añadido y su relevancia podrán ser diferentes para diferentes subgrupos de pacientes, en diferentes situaciones clínicas y frente a diferentes comparadores. Todas aquellas situaciones que puedan resultar de ayuda a la decisión posterior tendrán que ser analizadas.
Artículo 9. Aspectos específicos de la evaluación clínica de los medicamentos.
El informe de evaluación clínica de los medicamentos considerará la información siguiente:
a) Los datos clínicos sobre seguridad y eficacia incluidos en el expediente presentado a la agencia reguladora que corresponda;
b) todas las informaciones, datos, análisis y otros elementos de evidencia actualizados, publicados y no publicados, así como los informes y protocolos de los ensayos y los planes de análisis correspondientes a estudios del medicamento de los que el desarrollador de tecnologías sanitarias hubiera sido la entidad promotora, así como toda la información disponible sobre estudios del medicamento, en curso o suspendidos, de los que el desarrollador de tecnologías sanitarias sea la entidad promotora o en los que este tenga algún tipo de participación financiera, y la información correspondiente a estudios realizados por terceros, si se halla disponible, que sea pertinente para el ámbito de evaluación, incluidos los informes y protocolos de los ensayos clínicos, si están disponibles para el desarrollador de tecnologías sanitarias;
c) la información sobre estudios basados en registros;
d) si un medicamento ha sido objeto de una consulta científica, la explicación del desarrollador del mismo sobre toda desviación respecto de los medios de prueba recomendados;
e) la caracterización de la enfermedad o condición clínica que debe tratarse, incluida la población de pacientes destinataria;
f) la caracterización del medicamento objeto de evaluación;
g) la descripción de los métodos utilizados por el desarrollador del medicamento al elaborar el contenido del expediente;
h) los resultados de consultas para obtener información;
i) las características de los estudios que se adjunten;
j) los resultados sobre la eficacia y la seguridad de la intervención objeto de evaluación y del comparador;
k) los informes de evaluación sobre el medicamento cuando haya sido objeto de una evaluación clínica conjunta en el marco del Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021. En tal caso, el informe europeo constituirá la base de la evaluación clínica nacional y no será necesario presentar nuevamente la documentación prevista en los apartados a) a j), salvo que, de forma excepcional, se considere necesario realizar análisis adicionales relacionados con la población, intervenciones, comparadores o resultados en salud para su adecuada aplicación en el Sistema Nacional de Salud. En ningún caso se solicitarán al desarrollador datos, análisis u otros elementos de prueba que hayan sido presentados a escala de la Unión Europea.
Artículo 10. Aspectos específicos de la evaluación clínica de los productos sanitarios.
El informe de evaluación clínica de los productos sanitarios considerará la información siguiente:
a) La documentación presentada al organismo notificado de la evaluación clínica del fabricante con arreglo al anexo II, punto 6.1, letras c) y d), del Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017;
b) el dictamen científico emitido por los paneles de personas expertas pertinentes en el marco del procedimiento de consulta de la evaluación clínica, siempre que sea pertinente;
c) todas las informaciones, datos, análisis y otros elementos de evidencia actualizados, publicados y no publicados, así como los informes y protocolos de los ensayos clínicos y los planes de análisis correspondientes a estudios del producto sanitario de los que el desarrollador de tecnologías sanitarias hubiera sido la entidad promotora, así como toda la información disponible sobre estudios del producto sanitario, en curso o suspendidos, de los que el desarrollador de tecnologías sanitarias sea la entidad promotora o en los que este tenga algún tipo de participación financiera, y la información correspondiente a estudios clínicos y estudios observacionales realizados por terceros, si se halla disponible, que sea pertinente para el ámbito de evaluación determinado, incluidos los informes y protocolos de los ensayos clínicos, si están disponibles para el desarrollador de tecnologías sanitarias;
d) los informes de evaluación sobre la tecnología sanitaria objeto de una evaluación clínica conjunta, cuando sea apropiado;
e) los datos obrantes en registros que conciernen al producto sanitario e información sobre estudios basados en registros;
f) si un producto sanitario ha sido objeto de una consulta científica, una explicación del desarrollador de tecnologías sanitarias sobre toda desviación respecto de los medios de prueba recomendados;
g) la caracterización de la enfermedad o condición clínica que debe tratarse, incluida la población de pacientes destinataria;
h) la caracterización del producto sanitario objeto de la evaluación, incluidas sus instrucciones de uso;
i) el tema de investigación del expediente, desarrollado en el expediente de presentación, que refleje el ámbito de la evaluación determinado con arreglo al artículo 8.2 de este real decreto;
j) la descripción de los métodos utilizados por el desarrollador de tecnologías sanitarias al desarrollar el contenido del expediente;
k) los resultados de consultas para obtener información;
l) las características de los estudios que se adjunten;
m) los informes sobre los productos sanitarios cuando hayan sido objeto de una evaluación clínica conjunta en el marco del Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021. En tal caso, el informe europeo constituirá la base de la evaluación clínica nacional y no será necesario presentar nuevamente la documentación prevista en los apartados a) a l), salvo que, de forma excepcional, se considere necesario realizar análisis adicionales relacionados con la población, intervenciones, comparadores o resultados en salud para su adecuada aplicación en el Sistema Nacional de Salud. En ningún caso se solicitarán al desarrollador datos, análisis u otros elementos de prueba que hayan sido presentados a escala de la Unión Europea.
Artículo 11. Aspectos específicos de la evaluación clínica de los productos sanitarios para diagnóstico in vitro.
El informe de evaluación clínica de los productos sanitarios para diagnóstico in vitro considerará la información siguiente:
a) El informe de evaluación del funcionamiento del fabricante;
b) la documentación de la evaluación del funcionamiento del fabricante a la que se refiere el anexo II, punto 6.2, del Reglamento (UE) 2017/746 del Parlamento Europeo y del Consejo, de 5 de abril de 2017;
c) el dictamen científico emitido por los paneles de personas expertas pertinentes en el marco del procedimiento de consulta de la evaluación del funcionamiento, siempre que sea pertinente;
d) el informe del laboratorio de referencia de la Unión Europea, siempre que sea pertinente;
e) los informes sobre el producto sanitario para diagnóstico in vitro cuando haya sido objeto de una evaluación clínica conjunta en el marco del Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021. En tal caso, el informe europeo constituirá la base de la evaluación clínica nacional y no será necesario presentar nuevamente la documentación prevista en los apartados a) a d) salvo que, de forma excepcional, se considere necesario realizar análisis adicionales relacionados con la población, intervenciones, comparadores o resultados en salud para su adecuada aplicación en el Sistema Nacional de Salud. En ningún caso se solicitarán al desarrollador datos, análisis u otros elementos de prueba que hayan sido presentados a escala de la Unión Europea.
Artículo 12. Evaluación de los aspectos no clínicos.
1. La evaluación de los aspectos no clínicos incluirá el análisis económico y presupuestario, así como los aspectos éticos, organizativos, sociales, jurídicos, de género y ambientales. Se basará en la información proporcionada por las entidades desarrolladoras y, en lo que corresponda, en otras fuentes de información que se consideren pertinentes, de acuerdo con los criterios establecidos en las instrucciones y las directrices metodológicas no normativas.
2. La evaluación económica ofrecerá información útil para la toma de decisiones considerando el valor de la tecnología sanitaria en términos de efectividad relativa, teniendo en cuenta el valor social y el impacto sobre la calidad de vida relacionada con la salud de la tecnología sanitaria. Determinará la relación entre los resultados adicionales en salud que se espera que proporcione una nueva tecnología y los recursos adicionales que exige emplear, en principio, desde la perspectiva del Sistema Nacional de Salud, pero considerando otros aspectos cuando ello sea relevante. El objetivo, por tanto, será identificar la eficiencia de la nueva tecnología en comparación con las alternativas disponibles y será exigible, de acuerdo con las directrices no normativas de desarrollo, cuando las tecnologías evaluadas aleguen un valor clínico adicional sobre las alternativas existentes.
3. Además, siempre en el caso de los medicamentos y cuando proceda en el caso de otras tecnologías no farmacológicas, se realizará un análisis del impacto presupuestario desde la perspectiva del Sistema Nacional de Salud siguiendo también las instrucciones normativas de desarrollo aprobadas de acuerdo con lo establecido en el artículo 22 de este real decreto para la realización de este tipo de estudios.
4. Las Oficinas revisarán la calidad de los análisis económicos y presupuestarios presentados por el desarrollador y podrán requerirle la corrección y mejora de estos según los parámetros y criterios establecidos en las directrices no normativas de desarrollo aprobadas.
5. La revisión y la realización de estudios, así como el desarrollo de métodos, procedimientos y directrices de evaluación estarán sólidamente respaldados por los conocimientos científicos y las metodologías técnicas reconocidos por la comunidad científica y profesional y en línea con los estándares internacionales.
Artículo 13. Evaluación y protección de datos personales.
Todas las informaciones, análisis, datos y otros elementos de evidencia que se presenten o se elaboren en el contexto de las actividades recogidas en los artículos 7 a 12 de este real decreto, así como los sistemas de información y uso de datos y datos en vida real recogidos en el artículo 18 del mismo, que puedan ser considerados datos de carácter personal, ya se trate de categorías especiales de datos o no, se tratarán de forma anonimizada, y en su defecto, si no se puede cumplir con la finalidad de la ETS, los datos se tratarán de forma seudonimizada, de acuerdo con el Reglamento (UE) 2016/679 del Parlamento Europeo y del Consejo, de 27 de abril de 2016, relativo a la protección de las personas físicas en lo que respecta al tratamiento de datos personales y a la libre circulación de estos datos y por el que se deroga la Directiva 95/46/CE, y la Ley Orgánica 3/2018, de 5 de diciembre, de Protección de Datos Personales y garantía de los derechos digitales. Se considerará que es necesario tratar los datos de forma seudonimizada cuando sea necesario asegurar la trazabilidad temporal de los datos o la atribución a un mismo individuo de datos procedentes de distintas fuentes.
Artículo 14. Plazos de las evaluaciones.
1. Para el cómputo de los plazos, se considerará como fecha de inicio:
a) Para los medicamentos referidos en el apartado 1.a) del artículo 6 de este real decreto, la fecha en la que el desarrollador comunique su intención de comercializar en España, entendida como una comunicación expresa después de la opinión positiva del Comité de Medicamentos de Uso Humano de la Agencia Europea de Medicamentos o la concesión de código nacional por la AEMPS, lo que antes ocurra;
b) para los medicamentos referidos en el apartado 1.b) del artículo 6 de este real decreto, la concesión de código nacional por la AEMPS cuando se cumplan los criterios mencionados en ese apartado publicados por el Consejo o la fecha en la que se adopte la decisión de proceder a una evaluación por éste;
c) para los medicamentos referidos en el apartado 1.c) del artículo 6 de este real decreto, la fecha de la opinión positiva del Comité de Medicamentos de Uso Humano de la Agencia Europea de Medicamentos o la fecha de concesión de la autorización de la nueva indicación por la AEMPS siempre que se cumplan los criterios desarrollados y publicados por el Consejo de ETS;
d) para los productos sanitarios referidos en los apartados 1.d) y 1.e) del artículo 6 de este real decreto, la fecha de recepción de la documentación requerida al desarrollador;
e) para los medicamentos u otras tecnologías sanitarias referidos en los apartados 1.f) y 1.g) del artículo 6 de este real decreto, la fecha de acuerdo inicio por parte del Consejo de ETS.
2. Los plazos para las evaluaciones serán los siguientes:
a) Para la finalización del informe sobre los aspectos clínicos de los medicamentos referidos en los apartados 1.a), 1.b), 1.c) y 1.e) de este artículo, excepto reevaluaciones, setenta días desde la fecha de inicio y, en todo caso, no más de quince días tras la publicación del informe por la Comisión Europea cuando proceda.
b) Para la finalización del informe sobre los aspectos no clínicos de los medicamentos referidos en los apartados 1.a), 1.b), 1.c) y 1.e) de este artículo, excepto reevaluaciones, el plazo será de setenta días a partir de la recepción del informe sobre los aspectos clínicos.
c) Los plazos contemplados en los apartados a) y b) anteriores podrán solaparse siempre que la disponibilidad de aspectos parciales de la evaluación sobre los aspectos clínicos permita el inicio de la evaluación de los aspectos no clínicos.
d) Para la finalización del informe sobre los aspectos clínicos y no clínicos de las tecnologías sanitarias no farmacológicas, excepto reevaluaciones, 140 días.
e) Para las reevaluaciones, cincuenta días.
3. Estos plazos serán ampliados en veinticinco días si, en cualquier momento, durante la elaboración de los proyectos de informes, se considera que es necesario recabar otras especificaciones o aclaraciones, o informaciones, datos, análisis u otros elementos de evidencia adicionales para llevar a cabo la evaluación. El desarrollador dispondrá de diez días para aportar la información adicional solicitada desde el momento de dicha solicitud. El desarrollador podrá pedir, de forma justificada, una ampliación de cinco días para este plazo si el requerimiento de información implica un volumen de trabajo que se pueda considerar relevante.
4. Las Oficinas velarán porque pacientes y personas cuidadoras, personas expertas clínicas y otras personas expertas pertinentes se involucren a título individual o colectivamente, a través de las organizaciones representativas, en el proceso de evaluación, dándoles la oportunidad de hacer aportaciones a los proyectos de informe. Dichas aportaciones se presentarán dentro del marco y en los plazos establecidos que se determinen en las instrucciones y directrices referidas y aprobadas conforme a lo establecido en el artículo 22 de este real decreto sin extender los plazos referidos en los apartados 2 y 3 de este artículo.
5. Los informes de evaluación, antes de su finalización, se proporcionarán al desarrollador de tecnologías sanitarias para, en su caso, presentar alegaciones en un plazo máximo de diez días, conforme a lo establecido en el artículo 82 de la Ley 39/2015, de 1 de octubre, del Procedimiento Administrativo Común de las Administraciones Públicas, y lo referido en el artículo 11.5 del Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021.
6. Tras la recepción y examen de las observaciones aportadas de conformidad con este artículo, las Oficinas elaborarán los informes revisados y los presentarán al Grupo de adopción correspondiente.
Artículo 15. Efectos de las evaluaciones.
1. Las evaluaciones a las que hacen referencia los artículos 6 a 12 de este real decreto, así como los artículos 7 a 12 del Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021, no son vinculantes, constituyendo elementos que informan las decisiones de las administraciones, pero no constituyen la propia decisión ni esta está vinculada a aquellas en plazos ni en contenido.
2. Las evaluaciones de tecnologías sanitarias respetarán el principio de coherencia y no duplicarán las evaluaciones que ya hayan sido realizadas sin perjuicio de las competencias atribuidas a cada administración.
3. El resultado de una evaluación podrá estar condicionado a la resolución de las incertidumbres que hayan sido identificadas en el transcurso de la misma, pudiendo proponer el diseño de estudios que permitan generar la evidencia para solventar estas incertidumbres. La implementación de estos, en formato de estudios de monitorización, registros o programas piloto, dependerá de la decisión de los órganos decisores reglamentariamente establecidos.
4. Para minimizar las incertidumbres en el momento de la evaluación, se fomentará el diálogo temprano con las entidades desarrolladoras en el marco de las consultas científicas a que se hace referencia en el artículo 19 de este real decreto.
Artículo 16. Publicación de las evaluaciones.
1. Los informes de evaluación de las Oficinas serán públicos de acuerdo con el procedimiento que se establezca en las instrucciones referidas y aprobadas conforme a lo establecido en el artículo 22 de este real decreto.
2. Adicionalmente, la valoración final sobre la posición relativa de la tecnología sanitaria llevada a cabo por el Grupo de adopción, será publicada por el órgano competente del Ministerio de Sanidad como parte de un informe público de financiación en el caso de los medicamentos o en la forma que corresponda para otras tecnologías no farmacológicas.
3. Cualquiera de los informes mencionados que se publiquen tendrá suprimida toda información confidencial o que esté sujeta a derechos del desarrollador de la tecnología, sin que la confidencialidad impida la publicación de los actos de decisión de los órganos colegiados de asesoramiento técnico y científico del Ministerio de Sanidad.
Artículo 17. Actualización y revisión de las evaluaciones.
1. Cuando existan razones, entre otras, de cambio en la evidencia científica, cambio en las condiciones económicas, o de innovación incremental que pudieran hacer cambiar una decisión relativa a un medicamento o tecnología sanitaria no farmacológica, se podrá proponer al Consejo de ETS, a instancia de las partes interesadas que manifiesten un interés en el procedimiento, la revisión de la evaluación. Ello sin perjuicio de las previsiones existentes para la actualización de la cartera común de servicios del Sistema Nacional de Salud.
2. Con el fin de actualizar rápidamente la valoración final sobre la posición relativa de una determinada tecnología en función de las novedades y los avances científicos, cada actualización deberá tener en consideración otros informes anteriormente publicados.
3. Cuando la actualización de una evaluación pueda cambiar sustancialmente los resultados de otras evaluaciones clínicas o no clínicas anteriores, el Grupo de adopción podrá concluir en sentido diferente a la valoración anterior sobre la posición relativa e incluso recomendar la desinversión si procede o el desarrollo de programas piloto para su evaluación con datos en vida real.
Artículo 18. Sistemas de información, uso de datos y datos en vida real.
1. Se posibilitará e impulsará el uso de datos en salud para aprovechar el potencial que ofrece el intercambio, uso y reutilización de datos sanitarios en beneficio de los pacientes, la investigación, la innovación y la regulación dentro de un sistema coherente, fiable, eficiente y garantista, siguiendo el Reglamento (UE) 2025/327 del Parlamento Europeo y del Consejo, de 11 de febrero de 2025, relativo al Espacio Europeo de Datos de Salud, y por el que se modifican la Directiva 2011/24/UE y el Reglamento (UE) 2024/2847, así como el resto de normativa aplicable y el marco jurídico europeo en materia de utilización de datos, como es el Reglamento (UE) 2022/868 del Parlamento Europeo y del Consejo, de 30 de mayo de 2022, relativo a la gobernanza europea de datos, y el Reglamento (UE) 2023/2854 del Parlamento Europeo y del Consejo, de 13 de diciembre de 2023, sobre normas armonizadas para un acceso justo a los datos y su utilización, además de la correspondiente normativa nacional de desarrollo que corresponda.
El Espacio Nacional de Datos de Salud será un entorno de procesamiento seguro, conforme al Reglamento (UE) 2025/327 del Parlamento Europeo y del Consejo, de 11 de febrero de 2025, para su utilización por los organismos de acceso a datos de salud de España, conforme a los principios y garantías del marco normativo.
2. Las directrices metodológicas de carácter no normativo aprobadas por el Consejo de ETS incluirán situaciones y condiciones de uso de datos clínicos en vida real en relación con el objeto de este real decreto, respetando las directrices sobre interoperabilidad del dato sanitario y su calidad que establezca el Consejo Interterritorial del Sistema Nacional de Salud. Las condiciones sobre su disponibilidad se regirán por la normativa sectorial nacional y europea aplicable. Se procurará la coordinación con las directrices nacionales establecidas por la unidad de la Administración General del Estado competente en materia de fomento del uso datos entre administraciones públicas en lo que estas normas permitan.
3. El Ministerio de Sanidad garantizará sistemas de registro e información que permitan el intercambio de información entre los diferentes intervinientes en el proceso de ETS, así como evaluar y monitorizar los resultados del Sistema de ETS. Para la materialización de este uso secundario de los datos de salud se utilizarán las capacidades, servicios y entornos de tratamiento seguro provistos por el Espacio Nacional de Datos de Salud, haciendo efectivos los principios y garantías exigidos por el citado marco normativo.
CAPÍTULO IV
Otras actividades del Sistema para la evaluación de las tecnologías sanitarias
Artículo 19. Consultas científicas.
1. Las Oficinas podrán llevar a cabo consultas científicas, a solicitud del desarrollador, para intercambiar información con las entidades desarrolladoras de tecnologías sanitarias sobre sus planes de desarrollo para una tecnología sanitaria concreta, tanto en el ámbito nacional como al amparo de lo dispuesto en el Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, y teniendo en cuenta las especificidades de cada una de ellas.
2. Cuando la consulta científica incluyera aspectos relativos a las competencias de los órganos colegiados de decisión, las Oficinas involucrarán en la consulta científica al órgano competente del Ministerio de Sanidad.
3. Estas consultas estarán dirigidas a orientar el desarrollo, de acuerdo con los criterios por los que luego pueden ser evaluadas y que están definidos en la legislación de precio y financiación, de medicamentos o en la de incorporación a la cartera común de servicios de tecnologías sanitarias no farmacológicas, pudiendo cubrir cualquier aspecto de la evaluación de tecnologías sanitarias.
4. El Ministerio de Sanidad publicará el procedimiento para la solicitud, priorización, procedimiento, desarrollo y resultado de estas consultas científicas en las instrucciones normativas referidas y aprobadas conforme a lo establecido en el artículo 22.2 de este real decreto. El contenido del documento final de la consulta quedará reservado para las partes directamente implicadas en la misma, no pudiendo ser publicado o compartido con terceras partes en ningún momento.
5. Cuando su tecnología haya sido objeto de una consulta científica, el desarrollador lo hará constar en la documentación presentada conforme a los artículos 7 a 12 de este real decreto. En todo caso, el documento final de la consulta científica no dará lugar a ningún efecto jurídico para la administración competente, el Sistema de ETS o las entidades desarrolladoras de las mismas, sin perjuicio de que el mismo deba formar parte de los respectivos expedientes y ser tenido en cuenta a título informativo dentro de los procedimientos correspondientes.
6. Las consultas científicas conjuntas a las que hacen referencia los artículos 16 a 21 del Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021, no podrán ser repetidas a nivel nacional, salvo con el fin de complementarla o para abordar cuestiones de un contexto específico relacionadas con el Sistema de ETS. En este caso, la Oficina correspondiente informará de ello al Grupo de Coordinación sobre Evaluación de Tecnologías Sanitarias de los Estados miembros del Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021, a través de los medios previstos en el mismo.
7. Las Oficinas incluirán información resumida anonimizada, agregada y no confidencial sobre las consultas científicas en sus informes anuales y en sus páginas web. En todo caso, las consultas científicas se regirán por los principios de participación, transparencia y conflicto de interés que se recogen en los artículos 24, 25 y 26 de este real decreto.
8. Las consultas científicas podrán estar sujetas a una tasa o precio público, según corresponda, que se fijarán de acuerdo con la legislación vigente.
Artículo 20. Detección de tecnologías emergentes.
1. Las Oficinas llevarán a cabo acciones para la detección de tecnologías emergentes de las que se espere que hayan de tener repercusiones importantes para pacientes y personas cuidadoras, la prevención de deficiencias e intensificación de discapacidades, la salud pública o los sistemas de asistencia sanitaria. Estas acciones estarán coordinadas con las iniciativas que se realizarán a nivel europeo, que se describen en el artículo 22 del Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021.
2. Estas acciones estarán guiadas por la orientación estratégica del Consejo de ETS y se plasmarán en informes que abordarán, en particular, las repercusiones clínicas estimadas y las posibles consecuencias organizativas y financieras de las tecnologías sanitarias emergentes para el Sistema Nacional de Salud.
3. Las Oficinas cooperarán para ello con el órgano competente del Ministerio de Sanidad y el Grupo de adopción, así como con otras agencias a nivel nacional o internacional, basándose en los informes científicos o las iniciativas existentes sobre tecnologías sanitarias emergentes y la información de fuentes pertinentes, incluidas las siguientes:
a) Registros de estudios clínicos e informes científicos;
b) la red reguladora europea de medicamentos o el grupo de coordinación de productos sanitarios;
c) las entidades desarrolladoras de tecnologías sanitarias sobre las tecnologías sanitarias que estén desarrollando;
d) las partes interesadas, incluyendo las organizaciones o asociaciones de pacientes, así como las personas con discapacidad, organizaciones de consumidores, organizaciones no gubernamentales del ámbito de la salud, entidades desarrolladoras de tecnologías sanitarias y profesionales sanitarios.
4. Las Oficinas pondrán esta información de manera completa a disposición del Consejo de ETS y del Grupo de adopción, y publicarán de manera resumida información anual con supresión de toda información confidencial.
5. Se podrán desarrollar programas pilotados para la implementación de una determinada tecnología en la práctica clínica teniendo en cuenta las incertidumbres, potenciales beneficios y metodología para controlar las primeras y maximizar los segundos. En todo caso, corresponderá a los órganos decisores reglamentariamente establecidos la implementación de dichos programas a través de los procedimientos legalmente establecidos.
Artículo 21. Cooperación voluntaria en la evaluación de las tecnologías sanitarias.
En relación con la cooperación voluntaria entre Estados miembros de la Unión Europea a la que se refiere el artículo 23 del Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021:
a) Corresponderá a las Oficinas la cooperación y el intercambio de información científica en los términos previstos en el Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021, en los aspectos relacionados con la evaluación y presentación de los informes, siempre de acuerdo con la política farmacéutica y de prestaciones del Ministerio de Sanidad;
b) corresponderá a la Dirección General de Cartera Común de Servicios del Sistema Nacional de Salud y Farmacia la cooperación y el intercambio de información sobre la adopción, toma de decisiones y política farmacéutica o de la prestación de servicios sanitarios, incluyendo el uso de tecnologías sanitarias después de su autorización por el procedimiento que les corresponda.
CAPÍTULO V
Garantías del Sistema para la evaluación de las tecnologías sanitarias
Artículo 22. Instrucciones normativas y directrices metodológicas para la evaluación de las tecnologías sanitarias en España.
1. El conjunto de instrucciones normativas y directrices metodológicas de carácter no normativo, incluyendo los modelos de expediente, necesarias para favorecer la ETS en España constituye el cuerpo documental de instrucciones para la ETS en España.
Dicho cuerpo documental será publicado por el Ministerio de Sanidad, en su portal de internet. La elaboración y modificación del cuerpo documental seguirá un procedimiento participativo, deliberativo y transparente que garantice la consideración de las opiniones de todas las partes interesadas. En todo caso, cuando proceda, serán coherentes con las publicadas en desarrollo del Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021.
2. Para los contenidos de carácter normativo, la aprobación corresponderá a la persona titular del Ministerio de Sanidad y se ajustará al procedimiento establecido para disposiciones de carácter general, con publicación en el «Boletín Oficial del Estado».
Sin perjuicio del desarrollo y publicación de otras, las instrucciones referidas en los artículos 7, 12, 14, 16, 19, 23 y 26 de este real decreto serán aprobadas mediante orden ministerial de la persona titular del Ministerio de Sanidad.
3. El Consejo de ETS ejercerá la orientación estratégica, la coordinación y la propuesta de aprobación del cuerpo documental de instrucciones para la ETS en España. Las directrices metodológicas que no contengan reglas procedimentales no generen efectos obligatorios para terceros o tengan consecuencias procedimentales negativas, podrán ser aprobadas por el Consejo de ETS y publicadas en el portal de internet del Ministerio.
Artículo 23. Obligaciones de las entidades desarrolladoras de tecnologías sanitarias y consecuencias del incumplimiento.
1. El desarrollador está obligado a presentar el modelo de expediente establecido en las instrucciones normativas referidas y aprobadas conforme a lo establecido en el artículo 22.2 de este real decreto y los anexos del Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021, bien cuando se cumpla alguno de los hitos mencionados en el artículo 14 de este real decreto, apartados 1.a),
1.b) o 1.c), bien a solicitud de las Oficinas en los casos mencionados en el artículo 14, apartados 1.d) y 1.e) de este real decreto.
En este segundo caso, la Oficina correspondiente informará al desarrollador de tecnologías sanitarias del ámbito de la evaluación y solicitará la presentación de un expediente que contenga informaciones, datos, análisis y otros elementos de evidencia completos y actualizados tanto sobre los aspectos clínicos como no clínicos.
La presentación del expediente para la evaluación en el Sistema de ETS se entiende sin perjuicio de la documentación de inicio de la solicitud de financiación y precio, que deba ser presentada, cuando proceda, ante el órgano competente del Ministerio de Sanidad.
2. Si el desarrollador desiste de presentar esta documentación o propone iniciar la evaluación en otro momento, lo comunicará o solicitará por escrito indicando los motivos.
3. Si el expediente presentado no cumple con los requisitos establecidos en las instrucciones referidas en el artículo 22 de este real decreto, el desarrollador deberá corregirlo en el plazo de diez días. Este plazo podrá ser ampliado hasta cinco días a petición del desarrollador cuando la aportación de los documentos requeridos presente dificultades especiales.
4. Si el desarrollador no presenta o no corrige el expediente, se suspenderá la evaluación y se harán constar los motivos en los informes públicos de actividad, informando de ello al desarrollador de la tecnología sanitaria.
5. El cómputo de los plazos a los que se refiere el artículo 14 de este real decreto quedará suspendido en cualquiera de las circunstancias enunciadas en los apartados 2, 3 y 4 hasta que la Oficina disponga de un expediente completo.
6. En caso de que se haya suspendido la evaluación clínica, si se recibe posteriormente informaciones, datos, análisis y otros elementos de evidencia correspondientes al expediente a que se refiere el apartado 1, se podrá reiniciar la evaluación durante el año en curso de conformidad con el procedimiento que figura en este real decreto.
7. Sin perjuicio de lo anterior, cuando se haya reiniciado una evaluación, se podrá solicitar al desarrollador de tecnologías sanitarias que presente actualizaciones de las informaciones, datos, análisis y otros elementos de evidencia inicialmente facilitados en los términos recogidos en el artículo 14.3 de este real decreto.
8. El desarrollador de una tecnología en evaluación estará obligado a aportar información sobre las fuentes de financiación públicas o procedentes de entidades sin ánimo de lucro, si existieran, así como la información en materia de costes de producción y desarrollo que sea necesaria para llevar a cabo el análisis económico, todo ello sin perjuicio de la información que deba ser aportada durante los procedimientos de financiación y precio.
9. Los expedientes de las entidades desarrolladoras deben cumplir, en la parte clínica, los requisitos siguientes:
a) Los elementos de evidencia presentados deberán ser completos en relación con los estudios y datos disponibles que puedan fundamentar la evaluación, teniendo en cuenta una perspectiva del ciclo de vida de la tecnología sanitaria;
b) los datos se habrán analizado utilizando métodos adecuados para responder a todas las cuestiones objeto de investigación en la evaluación;
c) la presentación de los datos estará bien estructurada y será transparente, de tal forma que permita una evaluación adecuada dentro de los plazos limitados disponibles;
d) incluirá la documentación correspondiente a la información presentada, de tal forma que se puedan verificar la exactitud de dicha información.
10. No obstante, para aquellas tecnologías de las que se disponga una evaluación clínica conjunta, la solicitud de información adicional al desarrollador se limitará a los aspectos del ámbito nacional que no hayan sido considerados en la evaluación conjunta europea de conformidad con lo previsto en el artículo 13 del Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021.
Artículo 24. Garantía de calidad.
1. El Sistema de ETS garantizará que el trabajo conjunto siga las recomendaciones estándar sobre la evaluación basada en la evidencia y se ejecute en tiempo oportuno.
2. Cada grupo establecerá procedimientos internos de trabajo que se revisarán periódicamente y estarán alineados con el marco de instrucciones normativas y directrices metodológicas no normativas. En el desarrollo de estos procedimientos, se tendrán en cuenta las especificidades de la tecnología sanitaria sobre la que trata.
3. El Ministerio de Sanidad y las Comunidades Autónomas establecerán un mecanismo, conjuntamente con el órgano responsable de la evaluación y comunicación en materia de calidad asistencial dentro del Ministerio de Sanidad, para que los informes de evaluación y decisiones del Sistema de ETS sean difundidos a los profesionales sanitarios que desempeñan labor asistencial.
Artículo 25. Garantías de participación de pacientes, consumidores y profesionales.
1. Se garantizará la participación sistemática de pacientes y personas cuidadoras, consumidores y profesionales sanitarios y no sanitarios, cuando proceda en el caso de las profesiones no sanitarias, en las actividades recogidas en este real decreto para incorporar su perspectiva en todas ellas.
2. El Ministerio de Sanidad publicará en su portal de internet la metodología para la participación de organizaciones o asociaciones de pacientes y personas cuidadoras, organizaciones de consumidores y organizaciones profesionales en las actividades que les correspondan, aprobada por la persona titular del Ministerio de Sanidad mediante orden ministerial a propuesta del Consejo de ETS y con participación de las Oficinas y el Grupo de adopción.
3. Las organizaciones o asociaciones de pacientes y personas cuidadoras, organizaciones de consumidores y organizaciones profesionales deberán cumplir los siguientes criterios:
a) Tener objetivos claramente definidos,
b) tener como parte de sus actividades un interés específico en el área de trabajo,
c) representar los intereses de pacientes y personas cuidadoras, consumidores o profesionales, según corresponda,
d) tener órganos de gobierno elegidos por sus miembros, y
e) actuar con transparencia, incluyendo sus fuentes de financiación, tanto públicas como privadas.
4. Para concretar la participación individual en las evaluaciones o grupos de trabajo de las personas enumeradas en el apartado 1, se solicitará a las organizaciones o asociaciones relacionadas con el tema a abordar en cada momento que designen a una persona experta.
Tanto estas como cualquier otra persona que pudiera participar adicionalmente en estas actividades como persona experta externa, deberán cumplir los siguientes requisitos:
a) Firmar una declaración de elegibilidad que incluirá tanto aspectos relacionados con la organización proponente, si procede, como aspectos individuales que acrediten el perfil o implicación en el tema;
b) firmar los documentos de declaración de conflicto de interés y confidencialidad sobre la información a tratar a que se hace referencia en el artículo 26 de este real decreto.
En caso de que alguna de las personas participantes haya declarado un conflicto de interés en relación con un asunto concreto, la vocalía en representación de las organizaciones o asociaciones de pacientes podrá participar en la correspondiente reunión con voz, pero sin voto.
5. Todas las estructuras que forman parte del Sistema de ETS garantizarán la gestión de los aspectos enumerados en los apartados 3 y 4 de cara a la participación de cada uno de los agentes en las diferentes actividades.
6. El Ministerio de Sanidad mantendrá un registro oficial, público y transparente de organizaciones interesadas en formar parte de la evaluación de tecnologías que deberá contener, al menos la misma información que la publicada por la Comisión Europea respecto a la Red de partes interesadas de la ETS establecida en el Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021.
7. El Sistema de ETS favorecerá que se lleven a cabo actividades formativas básicas dirigidas a pacientes, consumidores y profesionales en el ámbito de sus competencias.
Artículo 26. Garantías de transparencia y conflicto de intereses.
1. Todas las personas que participen en el Sistema de ETS llevarán a cabo sus actividades de manera independiente, imparcial y transparente.
2. El listado de miembros de los grupos de trabajo, así como de los participantes en las reuniones y en las evaluaciones será público.
3. Las personas participantes en el Sistema de ETS no tendrán interés económico, profesional o de otro tipo en la industria de las entidades desarrolladoras de tecnologías sanitarias que pueda afectar a su independencia o imparcialidad.
A estos efectos, se considerará conflicto de interés la participación en actividades de asesoría científica, estratégica o técnica realizada para la industria de las entidades desarrolladoras de forma directa o indirecta, así como la participación en órganos de dirección, asesoramiento o financiación vinculados a dichas entidades.
Mediante orden ministerial de la persona titular del Ministerio de Sanidad se publicarán los documentos en los que se declaren los conflictos de interés definidos en este apartado, así como sus efectos de cara a la participación en las actividades de evaluación, y los periodos de tiempo en los que será efectiva dicha incompatibilidad.
4. Todas las personas implicadas harán una declaración de sus intereses económicos y de cualquier otra relación profesional, personal o institucional que pueda afectar a su independencia o imparcialidad, y la actualizarán anualmente o siempre que cambien las circunstancias declaradas. Revelarán cualquier otro hecho del que lleguen a tener conocimiento y del que sea razonable esperar, de buena fe, que suponga u origine un conflicto de intereses. La declaración de conflictos de interés será pública en los términos que establezca la normativa de desarrollo.
5. Todas las personas participantes declararán, antes de cada reunión, cualquier interés que pudiera considerarse perjudicial para su independencia o imparcialidad en relación con los puntos del orden del día.
6. Todos los participantes estarán sujetos a la obligación de confidencialidad, incluso después de haber cesado en sus funciones.
7. En todo caso, la publicidad sobre la participación de las personas en el Sistema de ETS se ajustará a lo dispuesto en el Reglamento (UE) 2016/679 del Parlamento Europeo y del Consejo, de 27 de abril de 2016, relativo a la protección de las personas físicas en lo que respecta al tratamiento de datos personales y a la libre circulación de estos datos y por el que se deroga la Directiva 95/46/CE, (en adelante, Reglamento general de protección de datos) y la Ley Orgánica 3/2018, de 5 de diciembre.
8. El tratamiento de estos datos por parte del Ministerio de Sanidad estará circunscrito al cumplimiento del artículo 6.1, letra c) del Reglamento general de protección de datos, siendo el necesario para el cumplimiento de las obligaciones contenidas en el artículo 4 del texto refundido de la Ley de garantías y uso racional de los medicamentos
y
productos
sanitarios, aprobado
por
el Real Decreto Legislativo 1/2015, de 24 de julio, y el artículo 5 del Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021.
9. Las actas de las reuniones de los grupos colegiados serán públicas eliminando las referencias a los aspectos que puedan ser confidenciales.
Artículo 27. Financiación del Sistema para la evaluación de las tecnologías sanitarias.
1. La financiación del Sistema de ETS podrá articularse a través de los instrumentos previstos en el ordenamiento jurídico, incluyendo, en su caso, los mecanismos previstos en la disposición adicional sexta del Real Decreto Legislativo 1/2015, de 24 de julio, por el que se aprueba el texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios, las correspondientes consignaciones en los Presupuestos Generales del Estado, o los sistemas basado en tasas o precios públicos establecidos conforme a la normativa aplicable.
2. La financiación abarcará todas las actividades previstas en este real decreto y estará ajustada a las disponibilidades presupuestarias existentes. En el segundo semestre del año el Consejo de ETS, teniendo presentes los programas de actividad anuales de los organismos encargados de la evaluación de los medicamentos y de las tecnologías sanitarias no farmacológicas aprobará una propuesta para el año siguiente que será elevada al Consejo Interterritorial del Sistema Nacional de Salud.
3. Cada uno de los órganos implicados y el Sistema de ETS rendirá cuentas anualmente al Consejo de ETS, y a tal efecto hará un análisis de la inversión realizada y de los resultados obtenidos.
Disposición adicional primera. Grupo de adopción de las tecnologías sanitarias.
1. El Grupo de adopción analizará, a partir de los informes de evaluación de las Oficinas y de cualquier otra información relevante, incluido el expediente de solicitud de inclusión en la prestación sanitaria, los elementos técnicos que resulten de aplicación a cada tecnología sanitaria. El análisis se articulará de manera diferenciada por ámbitos y, en ningún caso el Grupo de adopción sustituirá, condicionará ni cuestionará las competencias de los órganos decisores. El proceso de adopción se hará de acuerdo con el desarrollo reglamentario mediante orden de la persona titular del Ministerio de Sanidad para que sirva de base a los órganos competentes para la toma de decisión en relación con la inclusión, exclusión o modificación de las condiciones de uso en la prestación pública sanitaria.
2. El Grupo de adopción es un órgano colegiado adscrito a la Secretaría de Estado de Sanidad a través de la Dirección General de Cartera Común de Servicios del Sistema Nacional de Salud y Farmacia. Tiene dos estructuras, una para medicamentos y otra para las tecnologías sanitarias no farmacológicas. En su configuración de tecnologías no farmacológicas, en función de la diversidad de tecnologías a valorar, esta función puede ser asumida, en caso de ser solicitada por el órgano colegiado de toma de decisiones, por Comités, Ponencias o Grupos de Trabajo ya existentes y dependientes de la Comisión de Prestaciones, Aseguramiento y Financiación o de la Comisión de Salud Pública.
3. El Grupo de adopción en su configuración de medicamentos está formado por:
a) La persona titular de la Dirección General de Cartera Común de Servicios del Sistema Nacional de Salud y Farmacia, que asume la presidencia del Grupo de adopción;
b) la persona titular de la Subdirección General de Farmacia del Ministerio de Sanidad, que asume la secretaría del Grupo de adopción, con voz y voto;
c) una vocalía designada por la Subdirección General de Farmacia del Ministerio de Sanidad;
d) tres vocalías de la Administración General del Estado no pertenecientes al Ministerio de Sanidad, designadas, respectivamente, por el Ministerio de Hacienda, por el Ministerio de Economía, Comercio y Empresa, y por el Ministerio de Industria y Turismo;
e) una vocalía por cada una de las comunidades autónomas designadas a propuesta de sus respectivas Consejerías de Sanidad;
f) tres vocalías en representación de profesionales sanitarios, designadas por la Dirección General de Cartera Común de Servicios del Sistema Nacional de Salud y Farmacia, dos expertas clínicas y una experta en evaluación clínica y económica;
g) una vocalía especialista en economía de la salud, designada por la Dirección General de Cartera Común de Servicios del Sistema Nacional de Salud y Farmacia;
h) una vocalía en representación de las organizaciones o asociaciones de pacientes designada por la Dirección General de Cartera Común de Servicios del Sistema Nacional de Salud y Farmacia;
i) una vocalía en representación de las organizaciones de consumidores, designada por la Dirección General de Cartera Común de Servicios del Sistema Nacional de Salud y Farmacia a propuesta del Consejo de Consumidores y Usuarios.
En caso de ausencia, enfermedad u otra causa legal, las vocalías del Grupo de adopción en su configuración de medicamentos serán sustituidas temporalmente por las personas que designe la persona titular del órgano que propuso su nombramiento, hasta la reincorporación de la persona titular de la vocalía o la designación de una nueva.
4. El Grupo de adopción en su configuración de tecnologías sanitarias no farmacológicas está formado por:
a) La persona titular de la Dirección General de Cartera Común de Servicios del Sistema Nacional de Salud y Farmacia, que asume la presidencia del Grupo de adopción;
b) la persona titular de la Subdirección General de Cartera de Servicios del Sistema Nacional de Salud y Fondos de Compensación del Ministerio de Sanidad, que asume la secretaría del Grupo de adopción, con voz y voto;
c) una vocalía designada por la Subdirección General de Cartera de Servicios del Sistema Nacional de Salud y Fondos de Compensación del Ministerio de Sanidad;
d) una vocalía de la Secretaría General de Salud Digital, Información e Innovación del Sistema Nacional de Salud, con rango mínimo de subdirector o subdirectora general;
e) tres vocalías de la Administración General del Estado no pertenecientes al Ministerio de Sanidad, designadas, respectivamente, por el Ministerio de Hacienda, por el Ministerio de Economía, Comercio y Empresa, y por el Ministerio de Industria y Turismo;
f) una vocalía por cada una de las comunidades autónomas designada a propuesta de sus respectivas Consejerías de Sanidad;
g) tres vocalías en representación de profesionales sanitarios, designadas por la Dirección General de Cartera Común de Servicios del Sistema Nacional de Salud y Farmacia, dos expertas clínicas y una experta en evaluación clínica y económica;
h) una vocalía especialista en economía de la salud, designada por la Dirección General de Cartera Común de Servicios del Sistema Nacional de Salud y Farmacia;
i) una vocalía en representación de las organizaciones o asociaciones de pacientes designada por la Dirección General de Cartera Común de Servicios del Sistema Nacional de Salud y Farmacia;
j) una vocalía en representación de las organizaciones de consumidores, designada por la Dirección General de Cartera Común de Servicios del Sistema Nacional de Salud y Farmacia a propuesta del Consejo de Consumidores y Usuarios.
En caso de ausencia, enfermedad u otra causa legal, las vocalías del Grupo de adopción en su configuración de tecnologías sanitarias no farmacológicas serán sustituidas temporalmente por las personas que designe la persona titular del órgano que propuso su nombramiento, hasta la reincorporación de la persona titular de la vocalía o el nombramiento de una nueva.
5. La pertenencia al Grupo de adopción es incompatible con la participación en las evaluaciones, las actividades de las Oficinas o en los Comités mencionados en el artículo 4.5 de este real decreto. Salvo nombramiento en función del cargo o de los apartados 3.e) y 4.f) anteriores, también será incompatible con la participación en los órganos colegiados de toma de decisiones sobre precio, financiación pública o la incorporación a la cartera de servicios del Sistema Nacional de Salud.
6. Las personas que hayan participado en las evaluaciones por parte de las Oficinas y los técnicos correspondientes del órgano competente del Ministerio de Sanidad podrán participar en las reuniones del Grupo de adopción con voz, pero sin voto.
7. Para aspectos concretos, el Grupo de adopción de las tecnologías sanitarias podrá contar con la participación ad hoc de personas expertas, profesionales y pacientes, que podrán participar en las discusiones con voz, pero sin voto, especialmente cuando sea necesario contar con la opinión de subgrupos específicos. En este caso se garantizará que, como mínimo, se cuente con representación de la organización de ámbito estatal más representativa de las personas afectadas.
8. Las entidades desarrolladoras de tecnologías podrán tener audiencia con el Grupo de adopción de las tecnologías sanitarias para facilitar la discusión con todos los interlocutores.
9. El Grupo de adopción actuará, en principio, por consenso. Cuando no pueda alcanzarse un consenso, una decisión sobre la adopción requerirá el apoyo de una mayoría simple de las vocalías. Los resultados de las votaciones se harán constar en las actas de las reuniones del Grupo de adopción. Cuando se celebre una votación, las vocalías podrán solicitar que las opiniones divergentes se hagan constar en el acta de la reunión en la que se haya celebrado la votación.
10. Para cada una de sus configuraciones, las funciones del Grupo de adopción son:
a) Adoptar un reglamento interno de funcionamiento y actualizarlo cuando sea necesario; este reglamento contendrá previsiones sobre la periodicidad de reuniones;
b) facilitar la integración de los informes de evaluación proporcionados por las Oficinas para que lleguen a los órganos establecidos reglamentariamente para la toma de decisiones como una valoración final sobre la posición relativa de la tecnología sanitaria;
c) adoptar las fases detalladas del procedimiento para asegurar el cumplimiento de los plazos establecidos reglamentariamente;
d) garantizar que sean coherentes con los criterios de precio y financiación o de inclusión, exclusión y modificación en la cartera de servicios del Sistema Nacional de Salud desarrollados en la regulación pertinente;
e) garantizar que se cumplan los criterios de calidad, participación y transparencia a que se hace referencia en los artículos 24, 25 y 26 de este real decreto;
f) adoptar y presentar cada año al Consejo de ETS un informe anual, en el que se proporcionará información sobre el trabajo realizado el año natural anterior a su adopción;
g) proponer mecanismos o fórmulas para minimizar las incertidumbres identificadas durante el proceso de evaluación o de adopción de las tecnologías sanitarias.
Disposición adicional segunda. Funcionamiento de la RedETS.
La RedETS se regirá por lo dispuesto en la Orden SSI/1833/2013, de 2 de octubre, por la que se crea y regula el Consejo de la Red Española de Agencias de Evaluación de Tecnologías Sanitarias y Prestaciones del Sistema Nacional de Salud, para desarrollar las atribuciones que le confiere este real decreto como Oficina para la evaluación de las tecnologías sanitarias no farmacológicas, y mantendrá el procedimiento de elaboración del Plan anual de trabajo que tiene establecido.
Disposición adicional tercera. Plazo para la aprobación de las instrucciones normativas y publicación de las directrices metodológicas.
El plazo para la aprobación y publicación de la primera versión de las instrucciones normativas y directrices metodológicas de carácter no normativo a que se refiere el artículo 22 de este real decreto será de un año desde su entrada en vigor.
Disposición transitoria primera. Régimen transitorio relativo a la selección de pacientes, consumidores y profesionales para participar en los grupos de trabajo.
En tanto se desarrollan y publican las normas correspondientes para proponer, seleccionar, incorporar y hacer efectiva la participación en la ETS de organizaciones o asociaciones de pacientes y personas cuidadoras, organizaciones de consumidores y organizaciones profesionales, los representantes de cada uno de estos grupos serán nombrados por los organismos responsables del funcionamiento de los grupos de trabajo en los que van a participar. Estos nombramientos tendrán que ser realizados conforme a las normas una vez que sean adoptadas.
Disposición transitoria segunda. Régimen de aplicación transitorio.
1. A partir de la finalización del plazo indicado en la disposición adicional tercera, se procederá a una aplicación progresiva de lo dispuesto en este real decreto, de acuerdo con el calendario y fases que apruebe la Dirección General de Cartera Común de Servicios del Sistema Nacional de Salud y Farmacia mediante resolución publicada en el «Boletín Oficial del Estado», previo informe favorable de la AEMPS, que deberá pronunciarse sobre la suficiencia de los recursos humanos, materiales y tecnológicos necesarios para la ejecución de cada una de las fases previstas.
2. Durante el periodo transitorio comprendido entre la entrada en vigor del real decreto y la plena implementación del nuevo modelo, continuará siendo de aplicación el régimen jurídico anterior, en tanto no se oponga a lo establecido en este real decreto y hasta que cada fase de despliegue entre en vigor conforme al calendario previsto.
3. La persona titular del Ministerio de Sanidad podrá dictar cuantas disposiciones sean necesarias para la interpretación, aplicación y seguimiento del proceso de implementación progresiva regulado en esta disposición.
Disposición derogatoria única. Derogación normativa.
Quedan derogadas cuantas disposiciones de igual o inferior rango se opongan a lo establecido en este real decreto.
Disposición final primera. Título competencial.
Este real decreto se dicta al amparo de lo dispuesto en el artículo 149.1.16.ª de la Constitución Española, que atribuye al Estado competencia exclusiva en materia de bases y coordinación general de la sanidad y legislación sobre productos farmacéuticos.
Disposición final segunda. Facultad de desarrollo.
Se faculta a la persona titular del Ministerio de Sanidad para dictar las disposiciones necesarias para el desarrollo de este real decreto.
Disposición final tercera. Evaluación de las medidas previstas.
El Ministerio de Sanidad incluirá la evaluación de las principales medidas previstas en este real decreto en su Plan de Evaluación departamental, en los términos que se desarrollen de la Ley 27/2022, de 20 de diciembre, de institucionalización de la evaluación de políticas públicas en la Administración General del Estado, con el fin de contribuir a la mejora de la eficacia, eficiencia y calidad de la actuación pública.
Disposición final cuarta. Entrada en vigor.
Este real decreto entrará en vigor a los veinte días de su publicación en el «Boletín Oficial del Estado».
Dado el 27 de mayo de 2026.
FELIPE R.
La Ministra de Sanidad,
MÓNICA GARCÍA GÓMEZ
Las informaciones publicadas en Redacción Médica contienen afirmaciones, datos y declaraciones procedentes de instituciones oficiales y profesionales sanitarios. No obstante, ante cualquier duda relacionada con su salud, consulte con su especialista sanitario correspondiente.







 Andalucía
Andalucía  Cataluña
Cataluña  Madrid
Madrid  C. Valenciana
C. Valenciana  Galicia
Galicia  Castilla y León
Castilla y León  País Vasco
País Vasco  Canarias
Canarias  C-La Mancha
C-La Mancha  Murcia
Murcia  Aragón
Aragón  Extremadura
Extremadura  Asturias
Asturias  Baleares
Baleares  Navarra
Navarra  Cantabria
Cantabria  La Rioja
La Rioja 



