Investigadores estadounidenses y europeos publican un estudio en Nature Medicine en el que informan sobre una nueva opción de tratamiento único para pacientes con fibrosis quística. La tirosina alfa1 (Ta1) es una nueva terapia terapéutica basada en una única molécula que no sólo corrige defectos genéticos y de tejido, sino que también reduce significativamente la inflamación observada en pacientes con fibrosis quística.
"En este momento hay múltiples tratamientos para la fibrosis quística y, aunque estos han mejorado la esperanza de vida de forma espectacular, todavía hay sólo una vida útil de unos 40 años para los pacientes. Ningún tratamiento puede mantenerse solo", dice el coautor del trabajo Allan L. Goldstein, profesor emérito en Residencia de Bioquímica y Medicina Molecular de la Facultad de Medicina y Ciencias de la Salud de la Universidad George Washingon (GWU, por sus siglas en inglés), en Estados Unidos. "Desarrollamos un solo tratamiento que potencialmente puede corregir el defecto genético que causa la fibrosis quística y disminuir la inflamación que ocurre como resultado", afirma.
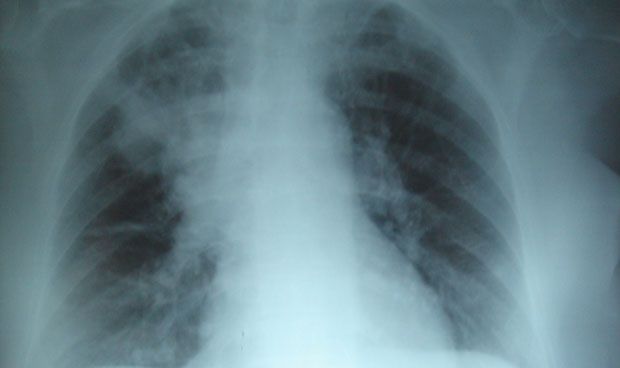
La fibrosis quística es una enfermedad genética que causa infecciones persistentes de pulmón y limita la capacidad de respirar con el tiempo y afecta a aproximadamente 70.000 personas en todo el mundo y 30.000 en Estados Unidos. La fibrosis quística es el resultado de mutaciones en el gen que codifica la proteína llamada regulador de conductancia transmembrana de fibrosis quística (CFTR, por sus siglas en inglés), que es importante para mantener la actividad del canal de cloruro que afecta al balance de sal y agua en los pulmones. Esta mutación tiene como resultado una proteína CFTR plegada erróneamente y su degradación prematura conduce a una permeabilidad perjudicial del cloruro y a una inflamación pulmonar persistente.
Múltiples defectos de tejido en los pulmones
Goldstein y los coautores Luigina Romani, en la Universidad de Perugia, y Enrico Garaci, en la Universidad de Roma 'San Raffaele', informan que Ta1, una versión sintética de un péptido natural aislado en primer lugar del timo, corrige los múltiples defectos de tejido encontrados en los pulmones e intestino delgado en un modelo de ratón de fibrosis quística, así como los defectos en el CFTR visto en las células aisladas de pacientes con fibrosis quística. Ta1 no sólo reduce significativamente la inflamación observada en la fibrosis quística, sino que también aumenta la maduración, la estabilidad y la actividad del CFTR.
Debido a esta doble acción, Ta1 ofrece un fuerte potencial para ser un agente terapéutico de una sola molécula para tratar y detener la progresión de la fibrosis quística. Goldstein y sus colegas primero aislaron y caracterizaron Ta1 como modificador de la respuesta biológica con potente actividad inmune terapéutica en 1979, una investigación que se realizó en gran parte en la Universidad George Washington.
Aunque el péptido se produce en pequeñas cantidades en varios tejidos linfoides y no linfoides periféricos, las concentraciones más altas de Ta1 se encuentran en el timo. Ta1, cuyo nombre comercial es Zadaxin, ha sido aprobado para uso clínico para más de 15 años en 35 países en el tratamiento de pacientes con infecciones virales, enfermedades de inmunodeficiencia, neoplasias malignas y VIH/sida. Aunque actualmente no está disponible en Estados Unidos, tiene un perfil de seguridad excelente y no induce los efectos secundarios y toxicidades comúnmente asociados con la mayoría de los agentes inmunomoduladores.
REGÍSTRATE GRATIS
PARA SEGUIR LEYENDO
¿Ya eres premium? Inicia sesión
Aviso importante
El usuario desde el que está intentando acceder a este contenido no está registrado como profesional autorizado para acceder a esta información. Esta noticia informa sobre novedades farmacológicas y, por ley, está reservada a profesionales de la salud habilitados para la prescripción o dispensación de medicamentos.
Volver a la portada de Redacción Médica
Las informaciones publicadas en Redacción Médica contienen afirmaciones, datos y declaraciones procedentes de instituciones oficiales y profesionales sanitarios. No obstante, ante cualquier duda relacionada con su salud, consulte con su especialista sanitario correspondiente.






 Andalucía
Andalucía  Cataluña
Cataluña  Madrid
Madrid  C. Valenciana
C. Valenciana  Galicia
Galicia  Castilla y León
Castilla y León  País Vasco
País Vasco  Canarias
Canarias  C-La Mancha
C-La Mancha  Murcia
Murcia  Aragón
Aragón  Extremadura
Extremadura  Asturias
Asturias  Baleares
Baleares  Navarra
Navarra  Cantabria
Cantabria  La Rioja
La Rioja 



